Grant - Summer 2010 - ALS — Michael Benatar

Michael Benatar, associate professor of neurology and epidemiology at Emory University in Atlanta, received an MDA grant totaling $525,000 to continue research into the early stage of FALS — familial, or inherited, ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease) — prior to symptom onset.
With funding from MDA, Benatar began his "pre-FALS" study in 2007, tracking a group of 30 people at risk for developing ALS because they harbor a mutation in the SOD1 gene, known to cause some forms of the disease. His team collected a series of physical, functional and neurological data over time in an effort to discern early biological markers ("biomarkers") of the disease process, and established a repository of biological samples collected from the study participants.
In Benatar's new follow-up study, the original group of 30 participants will expand to include presymptomatic individuals with mutations in other ALS susceptibility genes such as TDP43 and FUS.
Study aims include better definition of the presymptomatic stage of familial ALS, identification of environmental factors that might modify the age of disease onset among genetically susceptible individuals, continued development of biomarkers of early disease, and expansion of the existing collection of biological specimens.
Benatar's exploration into the presymptomatic phase of ALS could facilitate earlier recognition and diagnosis of both the familial and sporadic forms of the disease, which in turn could point the way toward therapeutics aimed at prevention or delay of ALS onset, as well as attempts to stop or slow the disease before it causes irreversible damage.
"MDA has shown great foresight in recognizing the importance of this study and the kind of long-term contribution that it can make to our understanding of the disease," Benatar said.
Funding for this MDA grant began August 1, 2010.
Grantee: ALS — Michael Benatar
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – CMT - Thien Nguyen, M.D., Ph.D.

MDA has awarded a research grant totaling $420,000 over three years to Thien Nguyen, assistant professor in the department of neurology at Johns Hopkins University School of Medicine in Baltimore. The new funds will help support Nguyen’s research into the breakdown of peripheral nerves (nervous tissue that connects the spinal cord with muscles and sensory organs) in Charcot-Marie-Tooth disease (CMT).
Degeneration of the axons (the long fibers that carry signals from nerve cell bodies to muscle) is a hallmark of type 1 CMT and is the primary determinant of the severity of symptoms in people with the disease. It follows that prevention of axonal degeneration should improve clinical outcomes for people with CMT1.
The same principle also should be applied to other types of CMT, Nguyen noted.
In previous studies, Nguyen and colleagues have determined that mutations in genes for myelin-making cells called "Schwann cells" can lead to mutant Schwann cells that may instruct axons to degenerate through certain specific signaling molecules that normally promote stability and survival, including one called myelin-associated glycoprotein (MAG). Study results out of Nguyen's lab have suggested that another signaling molecule, called netrin-1, may play an important role in axonal survival in CMT, making it a potential therapeutic target.
The investigators will determine whether increasing netrin-1 activity might prevent axonal degeneration in cell culture and in a research mouse model.
Findings from Nguyen’s work could reveal the potential therapeutic benefit of netrin-1 and lead to its development as a neuroprotective therapeutic for CMT.
"In theory, results from the project also could be expanded to axonal degeneration even without demyelination," Nguyen said. "Thus, there could be applicability to many of the neurodegenerative disorders covered by MDA."
Funding for this MDA grant began February 1, 2011.
Grantee: CMT - Thien Nguyen, M.D., Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – CMD - Shireen Lamande, Ph.D.
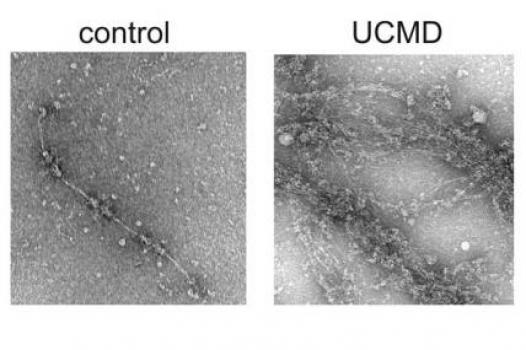
MDA has awarded a research grant totaling $251,596 over two years to Shireen Lamande, senior research fellow and group leader for muscular dystrophy research and musculoskeletal disorders at Murdoch Childrens Research Institute in Parkville, Victoria, Australia. The new funds will help support Lamande’s research into the identification of new genes responsible for two types of congenital muscular dystrophy (CMD), Bethlem myopathy and Ullrich congenital muscular dystrophy.
Lamande and colleagues plan to identify new genes responsible for collagen 6-related congenital muscular dystrophies, and determine how and why mutations associated with the genes cause muscle disease.
To date, the study team has screened 100 people with Bethlem myopathy and Ullrich CMD, and has found collagen 6 mutations in 62. The remaining 38 do not have collagen 6 mutations, an indication that other, as yet unidentified, genes also underlie these disorders.
Of the 38 individuals whose mutations were unknown, the investigators have identified new mutations in 11. They now plan to use a variety of methods to examine the newly uncovered genes, and identify those that are new muscular dystrophy candidate genes.
Results from Lamande’s work should improve the diagnostic process for individuals and families, and increase scientists’ understanding of muscle biology and disease.
Funding for this MDA grant began February 1, 2011.
Grantee: CMD - Shireen Lamande, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - ALS — Jean-Pierre Julien

MDA awarded a grant totaling $345,000 to Jean-Pierre Julien, professor at Laval University, Canada, for research into genetic variations in a protein called chromogranin B (CHGB) that has been shown to modify disease risk and hasten onset in a type of familial ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
In previous studies, Julien's group discovered that CHGB interacts with mutant, or flawed, forms of the superoxide dismutase 1 (SOD1) protein associated with SOD1-mediated familial, or inherited, ALS.
The team studied variations in the chromogranin B gene, which carries the instructions for the CHGB protein, in a large group of people with and without ALS. Results showed that people with ALS were twice as likely to have a common CHGB variant called P413L than those without the disease. The P413L CHGB variant also correlated with a 2.2-fold greater relative risk for development of ALS, and among those with the disease, onset of symptoms occurred an average 10 years earlier for those with the variant than for those without it.
In its new work, Julien's group will conduct experiments in cultured nerve cells and in a strain of mice engineered to carry the human CHGB variant to test the hypothesis that P413L increases vulnerability of motor neurons. The investigators also aim to determine whether the variant affects CHGB's interaction with mutant SOD1 and uncover the exact mechanism by which it increases risk of ALS and modifies disease onset.
"MDA funding is timely and needed," Julien said, "to determine how this particular genetic variant increases risk of ALS and hastens disease onset."
Funding for this MDA grant began August 1, 2010.
Grantee: ALS — Jean-Pierre Julien
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – CMD - Madeleine Durbeej-Hjalt, Ph.D.

MDA has awarded a research grant totaling $442,023 over three years to Madeleine Durbeej-Hjalt, a professor in muscle biology at Lund University (Sweden), Department of Experimental Medical Science. The grant will help support Durbeej-Hjalt's study of muscle-protein degradation processes in type 1A congenital muscular dystrophy (CMD1A).
CMD1A is caused by mutations in the gene for laminin alpha 2, a protein strand in a larger protein called merosin that connects muscle fibers to their surrounding tissue. Without the laminin alpha 2 strand, the normally three-stranded merosin protein can't perform this connective function and severe neuromuscular dysfunction results.
Although evidence indicates that loss of laminin alpha 2 leads to CMD1A, the underlying molecular mechanisms that cause muscle damage and loss in the disease remain unclear. Durbeej-Hjalt and colleagues hypothesize that muscle degeneration may be caused, at least in part, by increased destruction of muscle proteins through one or both of two major cellular protein degradation systems: the cellular "garbage can" known as the proteasome, or via the autophagic-lysosomal pathway, in which cell components are degraded and recycled through the cellular "recycle bin," or lysosome.
Durbeej-Hjalt's team has data indicating that activity in both protein degradation systems is increased when laminin alpha 2 is missing, and the investigators plan to test whether treatment with proteasome or autophagy inhibitors can block protein degradation and lead to improvement in muscle health.
Findings from Durbeej-Hjalt’s new study may generate important preclinical data that could lead to the development of therapies for people with CMD1A.
"I am extremely grateful for the MDA funding I have received since 2005," Durbeej-Hjalt said. "Without the MDA grants, my research group would not exist today."
Funding for this MDA grant began February 1, 2011.
Grantee: CMD - Madeleine Durbeej-Hjalt, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - ALS — Dena Jacob

Research scientist Dena Jacob at Thomas Jefferson University in Philadelphia, received an MDA grant totaling $180,000 for research into decreasing cells' resistance to therapeutic medications in ALS (amyotrophic lateral sclerosis, or Lou Gehrig’s disease).
Jacob plans to study a "transporter" protein called P-glycoprotein, or P-gp, which belongs to a class of "transporter" proteins that pump drugs out of cells. P-gp recognizes a broad range of drugs and normally is found in cells of the blood brain and blood spinal cord barriers and also in some affected brain tissue, limiting drug penetration.
"P-gp expression in the spinal cord itself may impart overt pharmacoresistance (drug resistance) to ALS-affected cells and account for the poor therapeutic effect observed in [a number of previous] ALS clinical trials," Jacob said.
Jacob's findings could lead to re-examination of many previously unsuccessful clinical drug trials and encourage the development of new, combined therapeutic strategies for ALS.
"I am extremely honored and excited to be a Development Grant recipient of the MDA, an organization whose efforts and support are essential for advancing our understanding of neuromuscular diseases," Jacob said. "This funding will enable me to complete important experiments that will help us identify improved therapeutic strategies to target the mouse model of ALS and, ultimately, humans with this disease."
Funding for this MDA grant began August 1, 2010.
Grantee: ALS — Dena Jacob
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - ALS — Daniela Zarnescu
MDA awarded $375,000 to Daniela Zarnescu, assistant professor in neuroscience at the University of Arizona in Tucson, Ariz., to conduct gene and drug discovery research in a drosophila fruit fly model that carries a mutation in the TDP43 gene associated with a genetic form of human ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
Alterations in TDP43 gene activity in the fruit fly model lead to symptoms that mimic those found in humans with the disease, including the death of nerve cells called motor neurons and the formation of abnormal clumps of cellular material, called inclusions, that contain TDP43 proteins.
Zarnescu's group will conduct genetic screening tests on the flies in order to identify other genes that interact with TDP43, some of which may be involved in disease causation or progression. The group also will screen drugs in search of one or more that can rescue the defects in the fruit fly model.
The work could lead to novel therapeutic targets and approaches for ALS.
"Recognizing the need to fund such projects is not only an important step to ensure important discoveries in the field, but it also shows that MDA is at the forefront of the effort towards identifying new therapies and a potential cure for ALS," Zarnescu said.
Funding for this MDA grant began August 1, 2010.
Grantee: ALS — Daniela Zarnescu
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - ALS — Daniel Offen

Daniel Offen, head of the neurology laboratory at Tel-Aviv University, Israel, received an MDA grant totaling $359,700 for research into a combined cell and gene therapy approach for ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
Offen's research team will engineer progenitor cells, a type of immature cell that forms muscle, to express various combinations of neurotrophic factors (proteins that support motor neuron health and which previously have been reported to be beneficial in rodent models of ALS). The investigators will inject mixtures of progenitor cells expressing different combinations of the neurotrophic factors into muscles in the SOD1 research mouse model of ALS and then monitor the course of the disease.
Behavioral, physical and biochemical survival indicators will be observed in order to analyze the effects of the stable activity of different growth factors on disease progression, extension of life span and quality of life in ALS. The experiments with the most promising results will then be duplicated using immature human muscle cells.
It's hoped the work will lead to a new cell and gene therapy strategy for treatment of human ALS.
"Past MDA support has enabled us to develop several approaches to isolate and propagate myogenic cells in cultures, some of which will be used in the present investigation," Offen said. "Our current MDA award will enable us to conduct the proposed project and hopefully to contribute to the treatment of ALS patients."
Funding for this MDA grant began August 1, 2010.
Grantee: ALS — Daniel Offen
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2010 - ALS — Brian Freibaum

MDA awarded $180,000 to research scientist Brian Freibaum at St. Jude Children's Research Hospital in Memphis, Tenn., for research into the mechanism by which toxic TDP43 protein leads to the development and progression of some forms of ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
Prior research has suggested that mutant (flawed) TDP43 protein is a critical component in the development of some forms of ALS, although the mechanism by which it works remains unknown. Freibaum has planned a three-tiered approach to uncover the protein's role in the disease.
First, Freibaum's team will test for toxicity in various tissues of fruit flies engineered to carry the human TDP43 gene. Using different mutant (flawed) and shortened forms of the protein is expected to help pinpoint what parts of the TDP43 protein are required for disease progression.
Next, the team will use human cell lines and mouse motor neurons (nerve cells that control muscle) to explore how TDP43 interacts with other proteins, where it normally localizes in the cell, and whether these locations are altered with mutant forms of the protein. Further experiments will be used to determine what other proteins interact with normal and flawed TDP43 protein.
Finally, the group will use the fruit fly model to perform genetic screens designed to explore how other proteins cooperate with mutant TDP43 to induce toxicity in motor neurons.
Greater understanding of the role TDP43 plays in ALS will help provide a more targeted approach to the development of new therapies.
"I am honored to be funded by such a prestigious organization that has a strong history in the advancement of neuromuscular disease research," Freibaum said. "This funding will be of great significance to our lab’s ALS research."
Funding for this MDA grant began August 1, 2010.
Grantee: ALS — Brian Freibaum
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2013 - SMA — Umrao Monani, Ph.D.

Umrao Monani, associate professor at Columbia University Medical Center in New York City, was awarded an MDA research grant totaling $300,000 over a period of three years to study how junctions between neurons and muscle are affected in spinal muscular atrophy (SMA).
SMA is due to a gene mutation that causes a deficiency in a protein calledSMN. Without it, the nerve cells that control muscles, called motor neurons, die off and muscles weaken. One of the first effects of lack of SMN is seen at the neuromuscular synapse, the place on the surface of the muscle where the motor neuron contacts it and sends its signal to control muscle contraction.
“We are interested in investigating the role of SMN in the formation, function, maintenance and repair of these structures,” says Monani. “We will use model mice, in which defects of the neuromuscular synapses become apparent when the SMN protein is depleted, to determine how SMN might shape synaptic structure and function.
"By examining molecular changes that occur normally in the motor neurons and muscle as the synapses mature, and by looking for differences when SMN is reduced, we hope to identify specific mediators of the SMA disease process. Understanding how low SMN levels translate into a disease that affects the motor system is not only of enormous scientific interest but is also likely to teach us how best to effectively treat SMA and other diseases of the motor neurons,” he says.
Funding for this MDA grant began August 1, 2013.
Grantee: SMA — Umrao Monani, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - SMA — Bennett Novitch, Ph.D.
Bennett Novitch, assistant professor of neurobiology at the University of California, Los Angeles, was awarded an MDA research grant totaling $300,000 over a period of three years to study the development of motor neurons that control respiration and their significance for spinal muscular atrophy (SMA).
SMA is due to the loss of motor neurons (nerve cells that control muscle activity), and in the most severe forms of SMA, the motor neurons controlling respiration are affected early in the disease.
“These observations raise the possibility that these respiratory defects might have a developmental origin [that is, they may arise in the developing embryo or fetus],” Novitch says, “resulting either from the improper formation of respiratory motor neurons, or their integration into functional motor circuits. This issue has been difficult to address, however, as our knowledge of the developmental origins of respiratory motor neurons and their assembly into circuits is incomplete.”
In this study, Novitch will study development of respiratory motor neurons and their organization into motor circuits that carry out both inspiration and expiration (breathing in and out). He also will study the gene networks that control this development. Better understanding of these networks, and the developmental processes they control, may lead to better understanding of how respiratory motor neurons are affected in SMA.
“Our study will provide new insights into the root causes of SMA and potentially lead to the discovery of new therapeutic targets. Moreover, by studying the process by which respiratory motor circuits are initially formed, we will gain vital information on how this activity may be recapitulated to rebuild damaged circuits to help patients maintain their ability to breathe independently,” he says.
Funding for this MDA grant began August 1, 2013.
Grantee: SMA — Bennett Novitch, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - SBMA/ALS — Constanza Cortes, Ph.D.

Constanza Cortes, a postdoctoral researcher at the University of California, San Diego, was awarded an MDA development grant totaling $177,410 over a period of three years to study the role of a cell recycling system inspinal-bulbar muscular atrophy (SBMA or Kennedy disease) andamyotrophic lateral sclerosis (ALS).
Both SBMA and ALS are characterized by the death of motor neurons, the nerve cells that control movement. Their causes are different, but in both diseases, there are defects in autophagy, a process cells use to break down and recycle large proteins and other cellular substances. Autophagy is critical for cell health, but relatively little is known about the process in neurons, Cortes says. She will be using both light microscopy and electron microscopy to examine how the function of one regulator of the process — a protein called TFEB — may be altered by interaction with the mutant protein that causes SBMA.
Working with cell culture, mouse models and cells from patients, Cortes says, “We expect to develop autophagy-intervention therapeutics and test these approaches in our mice, to determine the feasibility of manipulating autophagy as a therapeutic strategy for neuromuscular diseases. In this project, I hope to demonstrate the importance of neuronal autophagy for neuromuscular disease, and advance the field in ways that will benefit patients suffering from these debilitating disorders.”
Funding for this MDA grant began August 1, 2013.
Grantee: SBMA/ALS — Constanza Cortes, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS/SMA - Michael Miller, Ph.D.
MDA has awarded a research grant totaling $350,133 over three years to Michael Miller, associate professor in the department of biology at the University of Alabama, Birmingham. The grant will help support Miller's research into the fundamental molecular mechanisms responsible for the death of nerve cells called motor neurons in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease) and spinal muscular atrophy (SMA).
Using nematode (worm) and fly research models, Miller and colleagues have generated preliminary data suggesting that mutations in genes associated with familial (inherited) ALS and SMA disrupt a signaling mechanism that controls function of the cellular "energy factories" called mitochondria, which are necessary for motor neuron health.
Now, using a nematode model, the study team plans to combine biochemical and genetic manipulation with the ability to directly monitor changes in the worm’s cellular mitochondria as a means of uncovering the fundamental molecular mechanisms responsible for motor neuron death in ALS and SMA — mechanisms the team hypothesizes may influence mitochondrial function.
Miller expects the major impacts of his work will be the identification of mitochondria as the linchpin in neurodegenerative disorders including ALS and SMA, and the long list of drug targets his team expects to identify through its screening process.
"Funding by the MDA for this project is critically important," Miller said, "as without this support, the momentum gained over the past few years would come to a halt."
Funding for this MDA grant began February 1, 2011.
Grantee: ALS/SMA - Michael Miller, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2013 - SBMA — Albert La Spada, M.D., Ph.D.

Albert La Spada, professor of cellular and molecular medicine, neurosciences and pediatrics at the University of California, San Diego, was awarded an MDA research grant totaling $300,000 over a period of three years to study the causes of neurodegeneration (loss of nerve cells) inspinal-bulbar muscular atrophy (SBMA).
SBMA, also known as Kennedy disease, is an adult-onset neuromuscular disorder affecting men. It is caused by a mutation in the gene for theandrogen receptor protein, and it leads to the death of motor neurons, which are needed to control muscle contraction.
La Spada has created a mouse model and neuronal cell culture models of SBMA. He will now use these models to investigate how the SBMA-causing mutation leads to the death of motor neurons, focusing on changes in metabolism and impaired degradation of worn-out or defective proteins.
La Spada’s lab will test drugs that target a proposed regulatory factor and also will see whether blocking the erroneous androgen receptor gene instructions with compounds known as antisense oligonucleotides is beneficial. In previous work funded by MDA, La Spada’s lab has shown that directing therapies to muscle cells, rather than directly to the nerve cells that die in SBMA, can prevent the disease in mice.
“This exciting and unexpected finding represents an important step toward successful therapy development, as treatments that target muscle [instead of nerve cells] should be more straightforward to create, easier to test in patients and less likely to cause serious side effects,” La Spada says. “This key finding also suggests that a similar strategy is worth considering for related motor neuron diseases, such as spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS).”
Funding for this MDA grant began August 1, 2013.
Grantee: SBMA — Albert La Spada, M.D., Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS - Shanthini Sockanathan, Ph.D.

MDA has awarded a research grant totaling $396,000 over three years to Shanthini Sockanathan, associate professor of neuroscience at Johns Hopkins School of Medicine in Baltimore. The new funds will help support Sockanathan’s study of motor neuron (nerve cell) development in neurodegenerative diseases such as ALS (amyotrophic lateral sclerosis, or Lou Gehrig’s disease).
In previous work, Sockanathan and colleagues showed that a protein called GDE2 controls the production of spinal motor neurons through extracellular glycerophosphodiester phosphodiesterase (GDPD) activity.
More recently, the team discovered that GDE2 activity is itself regulated by a second protein, Prdx1. Prdx1 activates GDE2 by breaking a particular coupling called a "disulfide bond" inside GDE2 that normally inhibits GDE2 function. Thus, the disulfide bond within GDE2 acts a molecular switch that turns motor neuron production "on" or "off." By extension, the pathways that modify disulfide bond formation play pivotal roles in the control of motor neuron differentiation (maturation).
One question that emerges from this work is: How are active forms of Prdx1 generated to ensure efficient initiation of GDE2-dependent motor neuron differentiation?
Sockanathan's study team will test the hypothesis that inactive forms of Prdx1 are reactivated through separate pathways.
In addition, building off their recent finding of another Prdx molecule that is expressed with GDE2 and inhibits its activity, the team suggests the possibility that this mechanism regulates the initiation, extent and rate of motor neuron differentiation. The team will test this theory using a combination of different approaches.
Taken together, these studies are expected to provide insight into how the pathways that modify disulfide-bond formation intersect to control the onset and progression of motor neuron differentiation.
Funding for this MDA grant began February 1, 2011.
Grantee: ALS - Shanthini Sockanathan, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS - Jiou Wang, M.D., Ph.D.
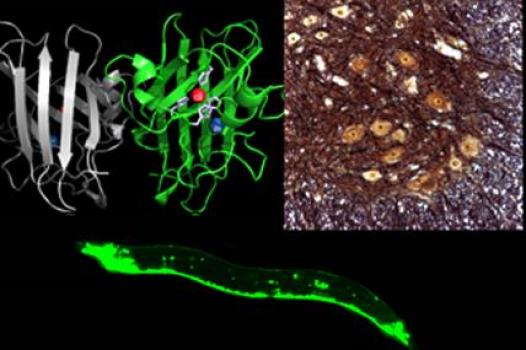
MDA has awarded a research grant totaling $330,000 over three years to Jiou Wang, assistant professor of biochemistry & molecular biology and neuroscience at Johns Hopkins University in Baltimore. The new funds will help support Wang’s study of the molecular mechanisms underlying ALS (amyotrophic lateral sclerosis, or Lou Gehrig’s disease).
"Genetic and protein discoveries in humans have opened the door to studying the disease in genetically-engineered animal models," Wang said. "The nematode, with its short lifespan and well-conserved nervous system, along with the availability of powerful genetic tools, provides a unique opportunity for dissecting the disease mechanisms of ALS."
With colleagues, Wang previously has established a nematode model of ALS that carries a mutation in the SOD1 gene (associated with the familial form of human ALS). In his new work, Wang and his study team plan to use the nematode model to compare the molecular mechanisms of toxicity in both the SOD1 protein and in another protein associated with ALS, TDP43. They aim to uncover the mechanisms by which the TDP43 and SOD1 disease genes trigger neurotoxicity, and then determine whether the same holds true in other mammals. It’s hoped in-depth analysis of these mechanisms will reveal pathways at which researchers can target effective therapeutics.
"Support from MDA is very special to us, as it allows us to continue our research into ALS," Wang said. "Since my graduate studies, I have focused my research on understanding the basic mechanisms of this devastating disease. Now, as an early-stage independent investigator, MDA support is critical in allowing me to continue to work on ALS."
Funding for this MDA grant began February 1, 2011.
Grantee: ALS - Jiou Wang, M.D., Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS - James Berry, M.D.

MDA has awarded a clinical research training grant totaling $180,000 to clinical research fellow James Berry at Massachusetts General Hospital (MGH) in Boston. The grant will support completion of a two-year fellowship during which Berry plans to study the effects of a drug called ISIS-333611 in familial, or inherited, ALS (amyotrophic lateral sclerosis, or Lou Gehrig’s disease).
Familial ALS accounts for 5 to 10 percent of all ALS cases. About 20 percent of those (1 to 2 percent of all ALS cases) are caused by a mutation in the SOD1 gene, which leads to production of abnormal SOD1 protein.
Berry is a member of the study team currently planning a phase 2 clinical trial of intrathecal (into the spinal canal) administration of the experimental drug ISIS-333611 (made by Isis Pharmaceuticals of Carlsbad, Calif.) in people with familial ALS. The trial will be a follow-on study to a phase 1 trial of the drug, currently under way, if results from that trial are favorable.
Previous studies of the drug in rats have shown that it is safe and tolerable, that it reduces mutant SOD1 RNA (the chemical step between DNA and protein synthesis) and protein levels, and slowed progression of the disease. It's thought that decreasing mutant SOD1 levels in humans may confer similar therapeutic benefits.
The planned phase 2 study will test ISIS-333611 over an extended period of time to assess its safety and tolerability. If the phase 2 trial is successful, a phase 3 trial may be planned to test efficacy of the drug.
"MDA funding is critical to my career development and my ability to begin my career as a clinical researcher in ALS," Berry said. "I am incredibly excited to have received this award, and I am looking forward to getting started on the project."
Funding for this MDA grant began February 1, 2011.
Grantee: ALS - James Berry, M.D.
Grant type: Clinical Research Training Grant
Award total:
Institution:
Country:
Grant - Summer 2013 - OPMD — Grace Pavlath, Ph.D.

Grace Pavlath, professor of pharmacology at Emory University in Atlanta, Ga., was awarded an MDA research grant totaling $100,000 over a period of one year to characterize a more accurate mouse model ofoculopharyngeal muscular dystrophy (OPMD).
OPMD is an adult-onset disease characterized by eyelid drooping and difficulties in swallowing, as well as weakness in the thighs and upper arms. The mutation responsible for the disease is found within a gene calledPABPN1, which regulates expression (activity) of other genes. In the most common form of the disease, called autosomal dominant OPMD, those who are affected carry one mutated copy of the gene and one normal copy.
However, Pavlath says, current mouse models of OPMD contain two normal alleles of PABPN1 in addition to one mutant allele of PABPN1, meaning there is a higher level of normal PABN1 protein made in these mice than in humans with the disease. “Thus, these mouse models do not accurately reflect the genotype of OPMD patients,” she says.
Now, Pavlath has created a mouse model that is closer to the human situation, permitting her to conduct experiments to better characterize the effects of the mutation in this model.
“These mice will be the first opportunity to accurately model this disease in mice and could provide a tool both for understanding how the disease affects muscle and for finding therapies to treat the disease,” she says.
Funding for this MDA grant began August 1, 2013.
Grantee: OPMD — Grace Pavlath, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS - François Berthod, Ph.D.

MDA has awarded a grant totaling $347,094 over three years to François Berthod, a professor in the department of surgery at Laval University in Quebec City, Quebec, Canada. The funds will help support Berthod's study of the underlying molecular mechanisms and disease process in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
ALS is characterized by the degeneration of motor neurons (nerve cells that control muscle movement) in the spinal cord and brain.
Berthod, who has a background in tissue engineering, previously has shown that human motor neurons can differentiate (mature) from a population of stem cells obtained from human skin.
Now, Berthod plans to develop a three-dimensional model of the human spinal cord using neural (nervous system) cells obtained from the tissues of people with ALS. The human tissue-engineered spinal cord (TESC) model is expected to mimic the human disease "in vitro," or in a laboratory setting. Its design will permit testing of the interactions of various neural cell types, including motor neurons and neuroprotective cells such as astrocytes, microglia and Schwann cells, as a means of determining the conditions that induce or aid motor neuron death in ALS.
Findings from Berthod's work are expected to improve understanding of the causes and progression of sporadic ALS.
"MDA funding over the years has been the only source of support for our research on amyotrophic lateral sclerosis," Berthod said, adding that it's been "the strongest encouragement to continue developing innovative in vitro models of this disease."
Funding for this MDA grant began February 1, 2011.
Grantee: ALS - François Berthod, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS - Ellen Barrett, Ph.D.

Ellen Barrett, professor of physiology and biophysics at the University of Miami (Florida) Miller School of Medicine was awarded an MDA grant totaling $297,102 over three years. The funds will support Barrett's study of the disease process and potential therapies in familial, or inherited, ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
It has been observed in some cases of ALS that motor nerve terminals (the part of the motor neuron, or nerve cell that conveys signals from the brain to muscles) deteriorate prior to the death of the motor neuron cell body.
In previous studies, Barrett and colleagues have demonstrated that motor nerve terminal health relies heavily on the proper function of cellular "energy factories" called mitochondria. The team also has found that mitochondrial function in motor nerve terminals is impaired in presymptomatic mice with an ALS-like disease, and that it worsens as the mice age and begin to show symptoms.
In her new work, Barrett will test various mitochondria-protective drugs in a research mouse model of ALS and then observe whether more motor terminals remain intact and functional in mice that receive treatment versus mice that do not.
Results from this work may point the way toward a therapeutic strategy involving a combination of treatments: some designed to protect mitochondria and preserve motor nerve terminals, and others targeted toward preservation of motor neuron cell bodies.
"MDA funding has been critical to research progress in our laboratory," Barrett said. "We have especially appreciated MDA’s (1) willingness to support basic as well as clinical research with relevance to neuromuscular diseases; (2) support for pilot projects investigating new research directions; and (3) excellent reviewers, who have always read our grant applications carefully and offered valuable suggestions."
Funding for this MDA grant began February 1, 2011.
Grantee: ALS - Ellen Barrett, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2013 - Muscular Dystrophies — Lee Sweeney, Ph.D.

Lee Sweeney, director of the Penn Center for Orphan Disease Research and Therapy at the University of Pennsylvania Perelman School of Medicine in Philadelphia, was awarded an MDA research grant totaling $278,286 over a period of three years to test whether a new treatment that affects muscle calcium can slow the damage to muscle tissue in several forms of muscular dystrophy.
A common characteristic of several types of muscular dystrophy is loss of regulation of calcium within muscle tissue. Calcium plays an important role in muscle contraction, and the inability to keep its level well-regulated reduces the force the muscle can generate, and contributes to muscle degeneration. Sweeney has worked on the development and testing of a peptide (a short chain of amino acids) called CT38, which helps correct the calcium handling defect in muscle. The peptide has been shown to be safe in humans, and will now be tested in models of Duchenne muscular dystrophy, Miyoshi myopathy and myotonic muscular dystrophy.
“We will give this peptide to these mouse models of human muscular dystrophies and observe whether the muscles from these mice can generate more force over time, and if the peptide prevents eventual destruction of the skeletal muscles,” Sweeney says.
“For all of the muscular dystrophies, it is clear that there is the possibility of developing multiple drugs to go after different aspects of each disease. By developing these multidrug therapies, we hope we can move toward stopping disease progression. We feel that CT38 has the potential to be one of the drugs in our arsenal that will combat the muscular dystrophies.”
Funding for this MDA grant began August 1, 2013.
Grantee: Muscular Dystrophies — Lee Sweeney, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS - Alysson Muotri, Ph.D.
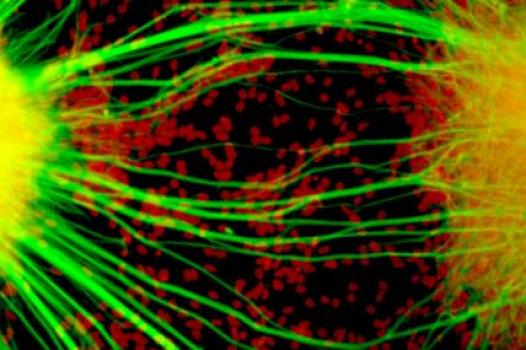
MDA has awarded a research grant totaling $362,466 over three years to Alysson Muotri, assistant professor at the University of California, San Diego in La Jolla. The new funds will help support Muotri’s generation of a new research model of ALS (amyotrophic lateral sclerosis, or Lou Gehrig’s disease).
Muotri and co-principal investigator Mayana Zatz, Ph.D., professor of genetics and director of the Human Genome Research Center in São Paulo, Brazil, plan to generate a human cellular model of ALS using cells taken from individuals with a particular form of the disease called ALS8.
ALS8 was first mapped in a Brazilian family, and is an autosomal dominant form of the disease, meaning an individual only has to receive one defective copy of the gene, from either parent, to get the disease. An individual with ALS8 has a 50 percent chance of transmitting the condition to each of their offspring.
Muotri noted that a critical need exists for new human cellular ALS models in which to study ALS, and suggested that recent advances in stem cell biology, including the availability of induced pluripotent stem cells (adult cells that are reprogrammed back to an immature stem-cell-like state) could help unravel the disease mechanisms underpinning ALS. These advances allow the DNA of people with ALS to be captured in an induced pluripotent stem cell line, from which cells can then be matured, isolated and evaluated for specific ALS-causing gene defects.
The new model will be made available to other research groups, and is expected to help determine biological indicators that can help scientists predict the disease course in individuals. It’s hoped the new model also will help make diagnosis easier with early-diagnostic tools and inspire new therapeutic strategies.
"The support of the MDA is crucial to moving the idea of modeling ALS disease using human motor neurons to real outcomes that can benefit patients," Muotri said.
Funding for this MDA grant began February 1, 2011.
Grantee: ALS - Alysson Muotri, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant – Winter 2011 – ALS - Adrian Israelson, Ph.D.

Update (April 1, 2015): Adrian Israelson is now at Ben-Gurion University of the Negev in Beer Sheva, Israel.
MDA has awarded a research development grant totaling $180,000 over three years to Adrian Israelson, a postdoctoral researcher at the University of California, San Diego in La Jolla, Calif. The funds will help support Israelson’s study of the underlying mechanisms governing motor neuron (nerve cell) death in SOD1-related familial ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
Israelson noted that mutant SOD1 has been shown to inappropriately associate with the cellular "energy factories," called mitochondria that are responsible for generating the power to run all cellular processes — but only in nervous system cells.
He previously has demonstrated in cell culture and in an ALS rat research model that mutant SOD1 binds directly to a key protein channel called VDAC1, which controls the import and export of chemicals required by mitochondria for power production. He also has shown that the misfolded portion of mutant SOD1 that is present in spinal cord mitochondria binds to the VDAC1 channel, inhibiting its conductance and compromising the energy supply for motor neurons.
Israelson's new work will focus on the ways in which mutant SOD1 protein associates with mitochondria, and whether and how that interaction affects the energy factories' function, possibly leading to motor neuron death. In addition the data may shed light on how modification of mutant SOD1 misfolding and the protein's association with mitochondria might affect mutant SOD1 toxicity.
Findings from these studies may provide valuable insight needed for development of therapies that either slow or prevent the degenerative process in ALS.
"Generous funding from MDA is very important," because of the Association's commitment to stopping rare diseases, Israelson said. "Thanks to the support of MDA, we’re able to keep investigating neuromuscular diseases that otherwise might go underfunded and unstudied."
Funding for this MDA grant began February 1, 2011.
Grantee: ALS - Adrian Israelson, Ph.D.
Grant type: Research Grant
Award total:
Institution:
Country:
Grant - Summer 2013 - Mito. Myopathy — Marilena D’Aurelio, Ph.D.

Marilena D’Aurelio, assistant research professor at Weill Medical College of Cornell University in New York City, was awarded an MDA research grant totaling $300,000 over a period of three years to test whether dietary supplementation can be therapeutic in mitochondrial myopathies.
Several muscle diseases are caused by genetic defects in cell structures called mitochondria. Mitochondria produce the energy used by muscle cells for all their functions, including contraction. In patients with some forms of mitochondrial myopathies, D’Aurelio has found specific impairments in the ability of cells to process the amino acid glutamine, which contributes to the energy deficit. She also has developed evidence that bypassing this process improves the survival of these cells. She will now study the glutamine impairment in a mouse model of mitochondrial myopathy, and explore the possibility that bypassing it with a dietary supplement can be therapeutic.
“In mitochondrial diseases, the energetic defects preferentially affect tissues with high-energy demand,” D’Aurelio says. “Brain, nerves, muscle and heart are often impaired in the same patient, presenting difficult and challenging conditions for diagnosis and therapy. Attempts to treat mitochondrial diseases have been disappointing thus far, due mostly to the lack of defined targets.”
Her work will test whether the impairment in glutamine processing may offer a viable therapeutic target.
Funding for this MDA grant began August 1, 2013.
Grantee: Mito. Myopathy — Marilena D’Aurelio, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - MMD — Thomas Cooper, M.D.
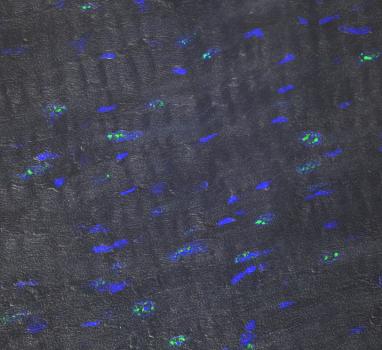
Thomas Cooper, professor of pathology and immunology at Baylor College of Medicine in Houston, Texas, was awarded an MDA research grant totaling $300,000 over a period of three years to develop “antisense” therapy approaches to treat type 1 myotonic muscular dystrophy (MMD1, also known as DM1).
MMD1 is caused by an expanded section of a gene, which in turn causes excess accumulation of a normal cellular molecule called RNA. The extra RNA then traps other molecules that are important for processing the genetic instructions that are used to make proteins. Cooper has created a mouse model of MMD1 in which he has begun to explore the details of these effects, and in which he will now begin to test a therapeutic strategy for the disease. He is developing antisense oligonucleotides — short RNA-like molecules that bind to the excess RNA, blocking its interactions with other substances or causing it to be broken down. A particular challenge is delivering the antisense oligonucleotides to the heart muscle, which is affected in MMD1; Cooper will be exploring how best to accomplish this in the mouse.
“There has been rapid progress in the use of antisense molecules as a potential therapy for myotonic dystrophy,” Cooper says. "In animal models, there has been some success delivering the therapy to skeletal muscles. We will focus on delivery of the antisense oligonucleotides to the heart.”
Funding for this MDA grant began August 1, 2013.
Grantee: MMD — Thomas Cooper, M.D.
Grant type:
Award total:
Institution:
Country:
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.

