Grant - Winter 2012 - SMA - Tomoyuki Awano, Ph.D.
Tomoyuki Awano, a postdoctoral research scientist in the department of pathology and cell biology at Columbia University Medical Center in New York, was awarded an MDA development grant (DG) totaling $180,000 over a period of three years to search for genes that modify the onset and disease course of spinal muscular atrophy (SMA). (MDA development grants are awarded to exceptional postdoctoral candidates who have the best chance of becoming independent researchers and future leaders of neuromuscular disease research.)
SMA is caused by the deletion of the SMN1 gene and a resulting deficiency of functional SMN protein. In humans, a nearly identical gene called SMN2 produces some partially functional SMN protein. Typically, the greater the number of copies a person with SMA has of the SMN2 gene, the less severe the disease.
But sometimes the SMN2 copy number doesn’t predict a person’s disease course.
This has been demonstrated in families, where multiple members are missing the SMN1 gene and have the same number of copies of SMN2, but whose SMA turns out drastically different. In previous work, Awano and colleagues have confirmed that this inconsistency is observed also in mouse models of SMA. A possible explanation is "modifier genes" that influence the disease course via the modulation of different biological pathways.
"Identifying modifier genes will not only reveal a potential target for cures, but will also shed light on the unknown disease mechanisms of SMA," Awano said.
Funding for this MDA grant began February 1, 2012.
Grantee: SMA - Tomoyuki Awano, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - SMA - Kentaro Sahashi, M.D., Ph.D.

Kentaro Sahashi, a postdoctoral research scientist at the Cold Spring Harbor Laboratory in Cold Spring Harbor, N.Y., was awarded an MDA development grant (DG) totaling $180,000 over a period of three years to study the roles of the SMN protein in spinal muscular atrophy (SMA). (MDA development grants are awarded to exceptional postdoctoral candidates who have the best chance of becoming independent researchers and future leaders of neuromuscular disease research.)
SMA is caused by mutations in the SMN1 gene, resulting in a deficiency of SMN protein. Humans have a closely-related SMN2 gene that also expresses functional SMN protein, but only in minute amounts due to differences in splicing, a cellular editing process that occurs during protein production.
In mice with an SMA-like disease, Sahashi and colleagues plan to use synthetic molecules called antisense oligonucleotides (ASOs) to influence and evaluate different SMN2 splicing patterns and determine whether they have potential therapeutic value.
The investigators plan to study the effects of ASOs in different tissues, administered via different delivery methods, and at different developmental stages. They expect to gain insight into the roles of the SMN protein in the SMA disease process, as well as an understanding of its normal functions in both the central nervous system and peripheral tissues.
"This in turn," Sahashi said, "will contribute to the ongoing development of targeted therapeutics and the establishment of a useful therapeutic time window."
Funding for this MDA grant began February 1, 2012.
Grantee: SMA - Kentaro Sahashi, M.D., Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - MMD — Nicholas Johnson, M.D.
Nicholas Johnson, assistant professor of neurology at the University of Utah in Salt Lake City, was awarded an MDA research grant totaling $375,000 over a period of three years to develop natural history data oncongenital-onset type 1 myotonic muscular dystrophy (MMD1, also known as DM1).
Congenital MMD1 is a severe form of myotonic dystrophy that begins in infancy. Because it is a rare condition, there is not as much known about the usual course of the disease as there is for adult-onset forms of myotonic dystrophy. Johnson will be conducting a “natural history” study to collect information on the most critical symptoms and how those symptoms change over time. Specifically, he will assess quality of life, cognition, speech, muscle strength and gastrointestinal symptoms in children with congenital MMD1 every year over a three-year period.
“This will allow for appropriate symptoms to be targeted in future treatment trials, as well as determining the age of children who will benefit the most from future treatments,” Johnson says.
“Recent data from preclinical models suggest antisense oligonucleotide therapy [a strategy for removing the harmful accumulation of RNA that causes the disease] may prove to be a very effective therapeutic approach for type 1 myotonic dystrophy, and human clinical trials are anticipated shortly. If antisense oligonucleotide therapy appears safe, there will be great urgency to extend clinical trials to children with myotonic dystrophy, particularly those most severely impacted by the disease,” he says. Therefore, collecting natural history data now will allow future trials to proceed more quickly.
Funding for this MDA grant began August 1, 2013.
Grantee: MMD — Nicholas Johnson, M.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - SMA - Christian Lorson, Ph.D.

MDA awarded a research grant totaling $381,582 over a period of three years to Christian Lorson, a professor in the departments of veterinary pathobiology, and molecular microbiology & immunology, at the University of Missouri in Columbia. The funds will help support Lorson’s research into targeting skeletal muscle as a therapeutic strategy in spinal muscular atrophy (SMA).
“Recently, several labs have shown the power of gene replacement — gene therapy — in SMA mice,” Lorson said. “Building upon these results, we will use a similar gene therapy strategy to investigate additional pathways that may provide insight into the SMA disease process, and also to identify novel therapeutic targets.”
Many therapeutic strategies under development for SMA involve replacement of the missing SMN protein; the aim of such strategies is to prevent loss of nerve cells called motor neurons.
Lorson and colleagues will use two different mouse models of SMA to test various gene therapy vectors (delivery vehicles) that instead target skeletal muscle. Their work is expected to determine whether such a therapy based on modulating activity of a non-SMN protein can reduce disease severity and extend survival.
Funding for this MDA grant began February 1, 2012.
Grantee: SMA - Christian Lorson, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2011 - ALS — Youngjin Lee, Ph.D.

Youngjin Lee, postdoctoral associate in the department of neurology at Johns Hopkins University School of Medicine in Baltimore was awarded an MDA development grant totaling $179,997 over three years. The funds will support Lee's study of the role of a protein called MCT-1 in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
One hypothesis to explain the death of motor neurons in ALS suggests that glial cells, whose job it is to provide nerve cells with nourishment and support, do not provide enough necessary energy components called substrates.
One of these substrates, lactate, is delivered to neurons by a type of transporter protein known as MCT-1. Lee’s research team has generated mice specifically designed to help elucidate the activity, regulation and function of MCTs and the protein’s role in neurodegeneration in ALS.
Favorable results could lead to new targets at which to aim therapeutic agents capable of preventing or slowing disease progression.
"Funding by MDA is 'absolutely crucial' for us to understand the balance of energy between motor neurons and their protective glial cells," Lee said, "and to find further therapeutic targets for preventing disease progression in ALS."
Funding for this MDA grant began August 1, 2011.
Grantee: ALS — Youngjin Lee, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - MMD — Michael Pape, Ph.D.

Michael Pape, president of Nymirum in Ann Arbor, Mich., was awarded an MDA research grant totaling $197,066 over a period of one year to develop drugs to treat type 1 myotonic muscular dystrophy (MMD1, also known as DM1).
MMD1 is due to a gene expansion, which causes a molecule called RNA to accumulate and aggregate (clump together). These RNA aggregates trap a protein called muscleblind, which normally regulates the formation of many other proteins. Loss of muscleblind accounts for many of the features of MMD. One strategy to treat the disease is to prevent the interaction of muscleblind and the RNA aggregates, by blocking the portion of the RNA (called a CUG repeat) where the two bind together.
“Several studies using small molecules and oligonucleotides show that CUG-binding molecules can inhibit muscleblind binding and alleviate the mutation-mediated dysfunctions,” Pape says. “While these studies provide proof of concept, none of the small molecules possesses the required efficacy [effectiveness] and/or drug-like properties for chronic treatment.”
Nymirum has discovered two sets of molecules that appear to have the potential to overcome these limitations. Nymirum will now pursue modifications to these molecules to increase their affinity for the expanded RNA and enhance their therapeutic potential.
Funding for this MDA grant began August 1, 2013.
Grantee: MMD — Michael Pape, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2012 - SBMA — Andrew Lieberman, M.D., Ph.D.

Andrew Lieberman, assistant professor of pathology at the University of Michigan Medical School in Ann Arbor, was awarded an MDA research grant totaling $405,000 over three years to study a new therapy approach for spinal-bulbar muscular atrophy (SBMA).
SBMA is an inherited degenerative disorder of lower motor neurons (nerve cells) that is caused by a mutation in the androgen receptor (AR) gene. The mutant protein causes toxicity associated with the male hormonetestosterone, resulting in muscle weakness and atrophy in men.
Previous work established that the mutant AR protein is the cause of this toxicity, suggesting that strategies aimed at more efficient degradation of the protein should diminish disease severity.
Lieberman and colleagues found that degradation of the AR protein is tightly controlled by cellular machinery consisting of the heat shock protein 70 (Hsp70).
The investigators now are testing genetic and pharmacologic (drug-related) methods to see whether increasing the stability of Hsp70 will help it more efficiently bind the mutant AR protein and promote its degradation.
“It is our expectation that this work will help define a new therapeutic approach to SBMA and other protein aggregation disorders where degradation of the mutant protein is controlled by Hsp70,” Lieberman said.
Funding for this MDA grant began Aug. 1, 2012.
Grantee: SBMA — Andrew Lieberman, M.D., Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - Pompe - Eric Sjoberg, Ph.D.

MDA awarded a research grant totaling $186,372 over a period of two years to Eric Sjoberg, principal scientist at Amicus Therapeutics in La Jolla, Calif. The funds will help support Sjoberg's research into immune system response associated with enzyme replacement therapy (ERT), the only approved treatment for Pompe disease (acid maltase deficiency or AMD).
In ERT, the acid maltase enzyme (GAA) is administered via intravenous infusion. The therapy has made a great difference in the lives of many people with Pompe disease, but challenges remain.
The majority of Pompe patients on ERT develop antibodies against GAA that can severely limit their ability to tolerate the drug, and also limit the ability of the drug to work, Sjoberg said. Evidence suggests that a specific genetic factor called "HLA type" in the patient population may be behind the unwanted immune response.
It's known that malformed or otherwise abnormal proteins are more likely to provoke an immune response than those with proper conformation (folding). GAA rapidly denatures in the neutral pH environment of plasma, the site in which the ERT is administered. This could be a possible cause of the ERT-mediated immune system response.
We have developed a specific pharmacological chaperone (a small molecule that selectively binds and stabilizes the target enzyme) for GAA, Sjoberg said. The molecule, AT2220, stabilizes the enzyme at neutral pH, Sjoberg said, noting that stabilizing GAA in plasma may mitigate the immune response associated with ERT treatment.
Sjoberg and colleagues plan to test ERT with and without AT2220 to determine whether a link exists between HLA types and ERT-mediated immune response. The investigators also will determine whether AT2220 can dampen ERT-related immune response.
Funding for this MDA grant began February 1, 2012.
Grantee: Pompe - Eric Sjoberg, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - MMD — Maurice Swanson, Ph.D.

Maurice Swanson, professor of molecular genetics and microbiology at the University of Florida College of Medicine in Gainesville, was awarded an MDA research grant totaling $300,000 over a period of three years to develop models to study sleep disturbance in types 1 and 2 myotonic muscular dystrophy (MMD1 and MMD2, also known as DM1 and DM2).
MMD1 and MMD2 are caused by a gene expansion that leads to loss of a protein called muscleblind. Muscleblind regulates the formation of many different proteins, only some of which affect muscle. People with MMD and their families have noted that one of the most debilitating aspects of this disease is hypersomnia or excessive daytime sleepiness, and animal models of the disease also display sleep disturbance. This suggests that loss of muscleblind in MMD likely affects the central nervous system's regulation of sleep.
Therefore, the goal of Swanson’s study is to define the molecular mechanisms underlying abnormal sleep regulation in MMD. He will develop mouse and cell models to study how normal circadian rhythms are altered by inhibition of muscleblind and related proteins, and identify the keycircadian cycle regulatory genes that are affected in MMD.
“This study should provide significant new insights into the cellular basis for abnormal sleep patterns” in MMD, he says, “and promotes the development of novel therapeutics to treat excessive daytime sleepiness.”
Funding for this MDA grant began August 1, 2013.
Grantee: MMD — Maurice Swanson, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2011 - ALS — Vasanthi Jayaraman, Ph.D.

MDA awarded $294,183 over three years to Vasanthi Jayaraman, an associate professor in the department of biochemistry and molecular biology at the University of Texas Health Science Center in Houston. The funds will help support Jayaraman’s study of the molecular mechanisms underlying motor neuron death in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
Jayaraman is particularly interested in a type of glutamate receptor known as calcium-permeable AMPA receptors. These cause toxicity and are thought to be a major trigger for selective motor neuron death and the resulting loss of muscle control in ALS.
Although AMPA receptor inhibitors have been shown to consistently decrease neuronal death in experimental models and increase survival time in a mouse model of ALS, they’ve not been tried in the clinic.
The challenge her research team has taken on, Jayaraman said, is to develop AMPA receptor antagonists that prevent activation of the calcium-permeable AMPA receptors without causing harmful side effects or affecting the function of other subtypes of glutamate receptors.
“With the funds provided by the Muscular Dystrophy Association,” Jayaraman said, “we are in a position to show the feasibility of these projects, which we believe will have high rewards in terms of being able to develop therapeutic agents toward ALS.”
Funding for this MDA grant began August 1, 2011.
Grantee: ALS — Vasanthi Jayaraman, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - Pompe - Darin Falk, Ph.D.
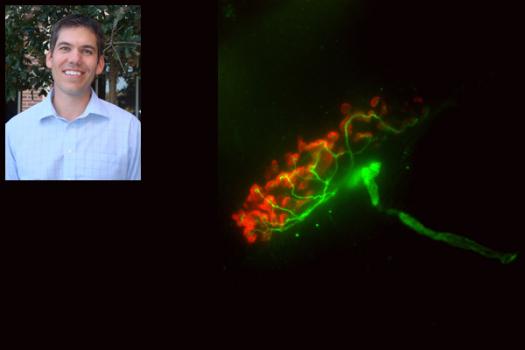
Darin Falk, a postdoctoral fellow in the department of pediatrics at the University of Florida in Gainesville, has received an MDA development grant (DG) totaling $179,846 over three years. The funds will help support Falk's research into the potential for gene therapy as a treatment forPompe disease (acid maltase deficiency or AMD). (MDA development grants are awarded to exceptional postdoctoral candidates who have the best chance of becoming independent researchers and future leaders of neuromuscular disease research.)
The current treatment for Pompe disease is enzyme replacement therapy (ERT), in which the acid maltase enzyme is administered to patients via intravenous infusion. ERT has dramatically changed the outcomes for people with Pompe disease but, Falk says, it "does not effectively reach and treat the disease component in the central nervous system."
In a Pompe research mouse model, Falk and colleagues will use the emptied-out shell of an adeno-associated virus (AAV) to deliver the gene for the acid maltase enzyme to the heart, skeletal muscles and spinal cord.
It’s hoped that the result will be sustained enzyme activity at appropriate levels in the targeted tissues.
Falk noted that while gene therapy already is a promising treatment strategy, further development and optimization of AAV delivery methods will allow scientists to more precisely target key tissues, resulting in improved outcomes for people with the disease.
Grantee: Pompe - Darin Falk, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2012 - MM - Mito. Myopathy — Michio Hirano, M.D.

MDA awarded a research grant totaling $398,532 over three years to Michio Hirano, professor of neurology and chief of the neuromuscular division at Columbia University Medical Center in New York. The funds will help support Hirano’s study of “molecular bypass therapy” for a mitochondrial myopathy called thymidine kinase 2 (TK2) deficiency.
Cellular “energy factories” called mitochondria generate the energy that fuels cells. Defects in mitochondria, which have their own DNA, are the causes of numerous human diseases that typically affect muscle and the brain.
One such disease, TK2 deficiency, usually begins in infancy. The disease also can manifest as adult-onset eye and limb muscle weakness. An enzyme (type of protein) called TK2 is required to synthesize the molecular building blocks of mitochondrial DNA.
With a previous MDA grant, Hirano and colleagues generated a research mouse model that has severely decreased TK2 activity and that develops muscle weakness similar to that of people with the infantile-onset form of TK2 deficiency.
In preliminary studies, administration of compounds to bypass the defective TK2 enzyme slowed the progression of the disease and doubled the life spans of the mice. Now, Hirano’s team is working to optimize the molecular bypass treatment in the TK2 mice.
Intensive research has revealed more than 200 genetically distinct mitochondrial diseases as well as novel therapeutic approaches for them, Hirano says. If successful, his new work could lead to the development of therapies for human TK2 deficiency and related diseases.
Funding for this MDA grant began Aug. 1, 2012.
Grantee: MM - Mito. Myopathy — Michio Hirano, M.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2011 - ALS — Raymond Grill, Ph.D.

MDA has awarded a research grant totaling $202,508 over a period of three years to Raymond Grill, assistant professor in the department of integrative biology and pharmacology at the University of Texas Health Science Center, Houston. The funds will support testing in the SOD1 mouse model of an experimental combination drug treatment in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
One process suspected to be heavily involved in ALS disease progression is inflammation, which can create a toxic environment and kill motor neurons.
Using the SOD1 ALS research mouse model, Grill's research team will test the hypothesis that a drug called Licofelone, which has completed phase 3 testing in humans, will enhance the ability of riluzole (Rilutek) to better penetrate the nervous system. (Rilutek is the only drug approved by the U.S. Food and Drug Administration for treatment of ALS.)
The researchers expect the combination treatment will reduce inflammation, protect motor function, rescue motor neurons and prolong survival in the SOD1 mice, and if favorable results are obtained, the group will attempt to "fast-track" the "two-part" treatment into clinical development.
"The support provided by the Muscular Dystrophy Association provides an invaluable opportunity for laboratories such as ours to enter this important field and both test and translate new ideas."
Funding for this MDA grant began August 1, 2011.
Grantee: ALS — Raymond Grill, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - Pompe - Celine Baligand, Ph.D.

MDA awarded a development grant (DG) totaling $170,873 over three years to Celine Baligand, a postdoctoral associate at the University of Florida College of Medicine in Gainesville. The funds will help support Baligand's research into the use of various imaging techniques to help determine the natural progression of Pompe disease (acid maltase deficiency or AMD). (MDA development grants are awarded to exceptional postdoctoral candidates who have the best chance of becoming independent researchers and future leaders of neuromuscular disease research.)
The current therapeutic approach for Pompe disease, enzyme replacement therapy or ERT, has improved outcomes for people with the disease. Alternatives to ERT, such as gene therapy, are under development.
Baligand and colleagues plan to develop tools, including magnetic resonance (MR) spectroscopy and other imaging techniques, for use in the noninvasive study of the natural course of Pompe disease in a mouse model. Such tools will facilitate the development of second- and third-generation treatment strategies for the disease.
"The development of MRI technology is crucial to improving the treatment of muscle diseases," Baligand said, noting that the development and validation of appropriate procedures for MRI could inform the preclinical testing of drugs and gene therapy in Pompe mouse models, potentially speeding the development of treatments for people with the disease.
Funding for this MDA grant began February 1, 2012.
Grantee: Pompe - Celine Baligand, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2012 - MMD — Thurman Wheeler, M.D.
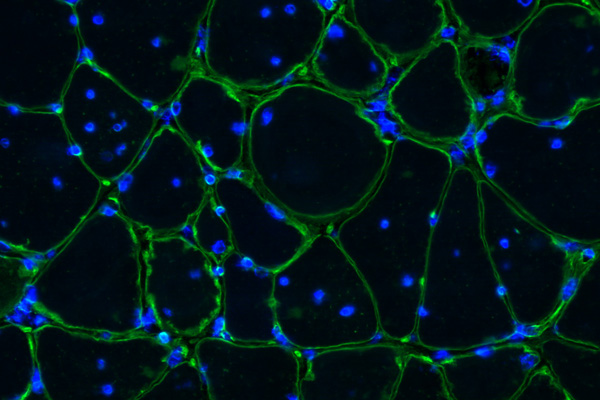
Thurman Wheeler, assistant professor in the department of neurology and Center for Neural Development & Disease at the University of Rochester, New York, was awarded an MDA research grant totaling $396,000 over three years to identify potential therapies aimed either at slowing down the progression of muscle degeneration or improving muscle health in type 1 myotonic muscular dystrophy (MMD1, or DM1).
The mechanism responsible for progressive muscle degeneration in MMD1 in humans is unknown.
Wheeler and colleagues have developed novel therapies that correct most aspects of myotonic dystrophy in mice with mild muscle degeneration. It’s unclear, however, whether the therapies will be as safe and effective in mice with advanced muscular dystrophy similar to MMD1 in humans.
Now the team is working with muscle cell cultures, muscle tissue and research mouse models of MMD1 to determine why progressive muscle degeneration occurs, and to identify therapeutic agents that slow progression of muscle degeneration or improve muscle pathology.
One of the therapeutic agents being tested by the team is antisense oligonucleotides (ASOs), administered by subcutaneous (under the skin) injection to mice with a disease resembling MMD1. The safety and efficacy of ASOs in mice with advanced muscle disease may help predict the safety and therapeutic response in people with MMD1 ahead of clinical trials.
Funding for this MDA grant began Aug. 1, 2012.
Grantee: MMD — Thurman Wheeler, M.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2011 - ALS — Junping Xin, Ph.D.

MDA has awarded a development grant totaling $180,000 over a period of three years to Junping Xin, research associate at the Neuroscience Institute, Loyola University Medical Center in Chicago, and Edward Hines Jr. Veterans Administration Hospital in Hines, Ill. The funds will help support Xin’s research into the possible effects of immune system dysfunction in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
In particular, Xin plans to study a specific type of immune system cell called the CD4+ T cell, which when dysregulated may lead to increased disease-related inflammation in people with ALS. It’s also known, however, that CD4+ T cells play an important role in supporting motor neuron survival.
Xin, and colleagues, plan to elucidate the mechanisms of the CD4+ T cell-mediated neuroprotection as well as the effects that result when the cells become dysfunctional.
A better understanding of both the normal and dysfunctional roles of CD4+ T cells should provide a better understanding of their possible use in new experimental therapies to treat ALS.
“MDA funding is critical to my being able to investigate these questions,” Xin said. “This grant funding will provide me with the chance to not only help people with ALS, but to develop an independent career in research medicine.”
Funding for this MDA grant began August 1, 2011.
Grantee: ALS — Junping Xin, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - Muscle Physiology - Elizabeth Chen, Ph.D.
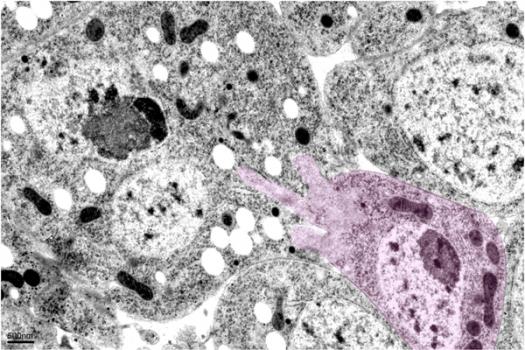
MDA has awarded a research grant totaling $321,489 over a period of three years to Elizabeth Chen, an associate professor at Johns Hopkins University School of Medicine in Baltimore. The funds will help support Chen's research into a mechanistic understanding of normal muscle physiology, which will inform potential therapeutic strategies aimed at treating various genetic and acquired degenerative muscle diseases.
Skeletal muscle is composed of large numbers of muscle fibers, each of which is the product of fusion between hundreds or even thousands of immature muscle cells called myoblasts. Myoblast fusion is not only important for skeletal muscle development, but also essential for stem-cell-based muscle regeneration.
Despite a large body of studies over several decades, the mechanisms underlying myoblast fusion in mammals remain poorly understood.
In this study, Chen and colleagues plan to characterize the role of a small type of protein called a GTPase during myoblast fusion in fruit fly (drosophila) muscle development and in a mouse model of muscle regeneration.
"While studies in Drosophila continue to provide novel insights into the molecular and cellular mechanism of myoblast fusion, the fundamental principles revealed by the Drosophila studies are now being tested in the mammalian systems," Chen said. "Such a cross-species approach will provide the basis for enhancing therapeutic efficacy in the treatment of a variety of muscle degenerative disease."
Funding for this MDA grant began Feb. 1, 2012.
Grantee: Muscle Physiology - Elizabeth Chen, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2012 - MMD — Charles Thornton, M.D.

Charles Thornton, professor of neurology at the University of Rochester in New York, was awarded an MDA research grant totaling $308,935 over three years. The funds will help support Thornton's work to expedite the development of effective treatments for type 1 myotonic muscular dystrophy (MMD, also known as DM).
MMD1 is caused by a repeat expansion mutation — an expanded section of DNA — in the DMPK gene, located on chromosome 19. Expanded DNA leads to the creation of expanded and toxic RNA, which causes problems for cells mainly because it traps and disables important proteins. Therefore, an important goal in MMD research is to free these cellular proteins — particularly one called muscleblind 1 or MBNL1 — so that they're able to perform their crucial functions.
One treatment approach that recently has emerged involves the use of small molecules to restore the activity of MBNL proteins, either by increasing their production or by blocking their interactions with toxic RNA. Another approach involves antisense oligonucleotides (AONs) that neutralize RNA toxicity or stimulate the clearance of toxic RNA.
"The model systems that are needed for preclinical testing of these respective approaches are somewhat different," Thornton explains. "Until now, the testing of small molecules has mainly been performed in HSALR transgenic mice, due to advantages of cost and efficiency; however, this model has limitations."
Thornton and colleagues are working to generate new mouse models that better reproduce the muscle weakness and degeneration that are characteristic in human MMD1. In addition, the team plans to design a model that will facilitate the testing of AONs.
Such mouse models could be used in the development and testing of potential new drugs to treat MMD.
Funding for this MDA grant began Aug. 1, 2012.
Grantee: MMD — Charles Thornton, M.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2013 - LGMD/Miyoshi Myopathy — Jyoti Jaiswal, Ph.D.

Jyoti Jaiswal, associate professor at George Washington University School of Medicine and Health Sciences and investigator at Children’s Research Institute in Washington, D.C., was awarded an MDA research grant totaling $300,000 over a period of three years to evaluate whether a new anti-inflammatory compound can reduce muscle damage in dysferlinopathies.
Dysferlinopathies are muscle diseases due to mutations in the dysferlin gene, an important muscle repair protein. They include limb-girdle muscular dystrophy 2B and Miyoshi myopathy. Jaiswal is studying patient cells and mouse models to determine whether a new compound, called VBP15, can improve muscle damage.
“Unlike some of the other muscular dystrophies where anti-inflammatory steroidal drugs are beneficial, these drugs are ineffective in dysferlinopathy patients,” Jaiswal says. “At present, there are no drugs available for dysferlinopathy, and the drugs being tested do not simultaneously address the poor muscle cell membrane repair and muscle inflammation — two of the key deficits common to dysferlinopathy patients.”
VBP15 improves the ability of muscle cells to heal damage to their outer membrane and is a potent anti-inflammatory agent that lacks the metabolic side effects associated with use of steroidal anti-inflammatory drugs. It is currently approaching clinical trials for other diseases.
“Establishing the preclinical benefits of VBP15 using dysferlinopathic mice could make this a viable drug-based therapy for dysferlinopathy patients,” Jaiswal says.
Funding for this MDA grant began August 1, 2013.
Grantee: LGMD/Miyoshi Myopathy — Jyoti Jaiswal, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2011 - ALS — Jasna Kriz, Ph.D.
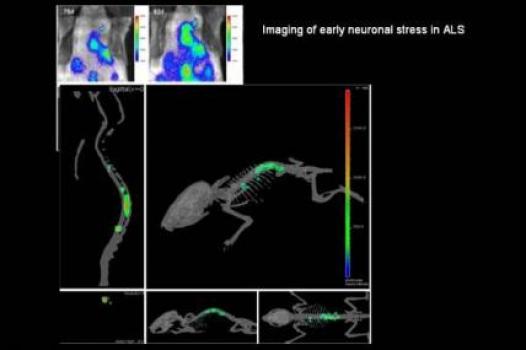
Jasna Kriz, associate professor in the department of psychiatry and neuroscience at Laval University, Quebec City, Canada, was awarded an MDA research grant totaling $445,086 over a period of three years to help refine and describe a new mouse model that will enable scientists to visualize different aspects of the disease process in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
Previously, Kriz and colleagues developed mouse models with bioluminescent and fluorescent genes (from fireflies) that allow researchers to visualize ALS-related events such as neuroinflammation and neuronal damage in the brains and spinal cords of living mice. They even were able to detect distinct and disease-specific signals linked to presymptomatic stages of the disease.
Now, Kriz plans to use the previously generated models to generate ALS imaging reporter mice, which will enable scientists to visualize, in live mice, different elements of the ALS disease process.
Once their characteristics are fully understood, the ALS reporter mice will represent a unique tool that will allow scientists to study the ALS disease process and real-time response to experimental therapies.
Favorable results are expected to lead to more efficient translation of experimental therapies to the clinic and new therapeutic strategies for ALS.
Funding for this MDA grant began August 1, 2011.
Grantee: ALS — Jasna Kriz, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - Muscle Physiology - Chris Weihl, M.D., Ph.D.

Chris Weihl, assistant professor of neurology at Washington University School of Medicine in St. Louis, was awarded an MDA grant totaling $397,064 over a period of three years. The funds will help support Weihl’s research into a process called autophagy in skeletal muscle. Data gleaned from Weihl’s studies may be applicable to a number of neuromuscular disorders including amyotrophic lateral sclerosis (ALS) and the muscular dystrophies; Weihl and colleagues will conduct their studies on a mouse model of myofibrillar myopathy (MFM).
Autophagy, which means "self-digestion," is a cellular cleanup and garbage-disposal system. Cells use it to degrade and destroy abnormal cellular or protein components that otherwise could lead to toxicity and cell death.
One hallmark in a number of neuromuscular diseases is the presence of protein clumps called aggregates or inclusions in affected tissues. It’s unknown whether facilitating the clearance or degradation of these inclusions is beneficial.
Weihl and colleagues plan to test FDA-approved drugs reported to enhance autophagy to determine whether protein degradation via autophagy is protective. The group will test the drugs in a newly developed mouse model of myofibrillar myopathy.
“These studies will answer the question of whether enhancing autophagy in protein-aggregate disorders is protective,” Weihl said.
Funding for this MDA grant began February 1, 2012.
Grantee: Muscle Physiology - Chris Weihl, M.D., Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Summer 2011 - ALS — Don Cleveland, Ph.D.

MDA has awarded a research grant totaling $429,983 over three years to Don Cleveland, departmental chair of cellular and molecular medicine; professor of medicine, neurosciences, and cellular and molecular medicine; and member of the Ludwig Institute for Cancer Research in La Jolla, Calif. The funds will help support Cleveland’s research into the connection between mitochondria and ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
Mitochondria, the intracellular compartments that consume oxygen to produce chemical fuel that supports the cell, have been implicated as targets for toxicity in the inherited, SOD1-associated forms of ALS.
It is not known, however, in which cell types of the nervous system mitochondrial damage occurs, whether the ensuing mitochondrial dysfunctions are a cause or a consequence of neuronal degeneration during the disease and if the effects are the same in SOD1-associated ALS caused by different SOD1 mutations.
Using mice that are genetic mimics of inherited, SOD1-related ALS, Cleveland intends to determine at what disease stage and in which cell types of the central nervous system mitochondrial dysfunction occurs, and whether rescue of individual mitochondrial functions or an increase in mitochondrial activity may alter the ALS disease course.
“This comparative analysis will, in principle, provide a test for whether mitochondria are a common target of toxicity,” Cleveland said.
Funding for this MDA grant began August 1, 2011.
Grantee: ALS — Don Cleveland, Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - MMD - Mani Mahadevan, M.D.

Mani Mahadevan, a professor at the University of Virginia in Charlottesville was awarded an MDA grant totaling $281,352 over a period of two years. The funds will help support Mahadevan's investigation into potential therapies for type 1 myotonic muscular dystrophy (MMD1, or DM1).
A key component of Mahadevan's new work is collaboration between the Mahadevan lab and Novartis through the Genomics Institute of the Novartis Research Foundation. Together, the two groups intend to identify compounds that correct defects in muscle tissue formation in DM1.
They will do this via a screening method that will evaluate approximately 850,000 compounds; promising candidates will then be evaluated in a mouse model to determine whether they lead to any improvement in the generation of mature muscles in DM1.
Compounds identified through the screening process may be used as starting points for further development of a DM1 therapy.
"The current state of translational research is very exciting," Mahadevan said. "A number of different approaches, including antisense oligos and small molecules are being developed and tested by labs around the world to try to develop therapies for myotonic dystrophy and have yielded promising results in mouse models."
Funding for this MDA grant began February 1, 2012.
Grantee: MMD - Mani Mahadevan, M.D.
Grant type:
Award total:
Institution:
Country:
Grant - Spring 2011 - ALS — Clotilde Lagier-Tourenne, M.D., Ph.D.

Clotilde Lagier-Tourenne, a postdoctoral fellow at the University of California, San Diego, in La Jolla, was awarded an MDA development grant totaling $180,000 over a period of three years to study the roles of two proteins, TDP43 and FUS, in ALS (amyotrophic lateral sclerosis, or Lou Gehrig's disease).
ALS-causing mutations in the genes for two RNA binding proteins, TDP43 and FUS, appear to cause disruption in the processing of RNA (the chemical step that directs protein synthesis).
In a research mouse model, Lagier-Tourenne and colleagues have identified RNAs that TDP43 normally binds to, as well as abnormalities in the processing of RNA that occur when TDP43 is depleted.
Similarly, in their new work, the researchers plan to determine the role of FUS in the regulation of RNA in the mouse central nervous system.
The team's proposed set of studies is expected to identify a set of alterations to normal RNA processing that will define a TDP43- and FUS-dependent ALS disease signature.
"I am really grateful for the support from MDA," Lagier-Tourenne said. "It represents strong encouragement at this stage of my career and will allow me to extend my research on the role of RNA processing in neurodegeneration."
Funding for this MDA grant began August 1, 2011.
Grantee: ALS — Clotilde Lagier-Tourenne, M.D., Ph.D.
Grant type:
Award total:
Institution:
Country:
Grant - Winter 2012 - MMD - Laura Ranum, Ph.D.

MDA awarded a research grant totaling $415,092 over a period of three years to Laura Ranum, professor of molecular genetics and microbiology at the University of Florida in Gainesville.
The funds will help support Ranum's research into the role of a phenomenon called Repeat Associated Non-ATG translation (RAN translation) in myotonic dystrophy (MMD, or DM1).
Ranum and colleagues have discovered a new mechanism, RAN translation, by which repeat sequences (series of repeated segments of DNA) direct protein synthesis in the absence of normal regulatory signals. Evidence suggests that RAN translation results in the production of unexpected mutant proteins in myotonic dystrophy.
In her new work, Ranum and colleagues plan to determine how many of these unexpected proteins are made in myotonic dystrophy, which cells in the body are able to make them, and what effects they have on the disease.
"We have demonstrated that RAN translation occurs in cell culture and in animal models of type 1 myotonic dystrophy, and in tissues from human patients," Ranum said. "We must now consider the effects that these newly discovered mutant proteins could have on the disease."
Funding for this MDA grant began February 1, 2012.
Grantee: MMD - Laura Ranum, Ph.D.
Grant type:
Award total:
Institution:
Country:
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.

