
FA: A Case of Impaired Ironworks

From childhood science classes, you may remember the periodic table listing all the known chemical elements. You might also recall, in approximately the center of the table, the symbol “Fe” signifying the metal iron.
Produced inside large stars in distant space, iron is the most common element on Earth. From earliest times, humans have used it in everything from paint pigments, cookware and tools to buildings, bridges and cars.
Longer still, humans have relied on iron as a biological workhorse, necessary for survival.
Iron-containing proteins are involved in a vast array of vitally important functions in the body including:
- formation of red blood cells;
- oxygen transport via blood and oxygen storage in muscle; and
- cellular energy production.
Distribution and storage of iron is carefully balanced and regulated by cells.
Too little can result in iron deficiency anemia, characterized by weakness and fatigue, and impairment to the immune system.
Too much iron, or iron overload, is toxic, with a broad range of effects that may include cell damage, organ damage (such as in the pancreas, heart and brain), metabolism problems and neurodegenerative disease.
The inability to properly manage iron lies at the heart of the neurodegenerative disease Friedreich’s ataxia (FA).
Finding the FA gene
In 1996, two teams of scientists — one led by then-MDA-grantee Massimo Pandolfo at Baylor College of Medicine in Houston, and the other by Michel Koenig in France — published their discovery of the underlying molecular cause of FA: an expanded sequence of extra DNA in a gene on chromosome 9. The gene carries instructions for production of the frataxin protein.
It wasn’t an easy find, but it was the crucial first step in providing researchers with targets at which to aim therapies for the disease.
“Finding the gene — it was a big, exciting time,” recalls Sanjay Bidichandani, MDA vice president for research, who was part of Pandolfo’s team on the gene identification project, with MDA support.
Bidichandani, also a former member of MDA’s Medical Advisory Committee (MAC), noted that pinpointing the gene was particularly difficult. While a majority of genetic diseases are caused by mutations in the “coding” regions, or exons, of genes (that is, the parts of the gene that carry instructions for protein synthesis), the defect responsible for FA turned out to reside in a “noncoding” region of the gene, or intron.
Genetic mutations in FA
Although two types of FA-causing mutations exist, by far the most common is a GAA trinucleotide repeat expansion — a region of DNA containing greater-than-normal numbers of the chemical phrase “GAA.”
Five to 30 GAA repeats are within normal range, but in people with FA the repeats number in the hundreds to thousands, with the larger repeat expansions generally correlating with earlier onset and greater disease severity.
Approximately 96 percent of people with FA have two GAA repeat expansions, one on each chromosome. The other roughly 4 percent possess two different mutations: an expansion on one chromosome, and a conventional gene mutation (most of which are truncations, or deletions) on the other.
Importantly, every individual with FA has at least one GAA expansion mutation, which has important implications for therapy development.
“There has never been a single Friedreich’s ataxia patient reported to date who has conventional, or point, mutations on both chromosomes,” Bidichandani says. “That’s important when it comes to developing therapeutic strategies, because it implies that treatments targeted to the repeat or its consequences should apply to every patient that exists.”
The location of the GAA expansion in an intron holds additional positive implications for treatment because the exons and the protein-making instructions they contain are left unaffected. The introns are removed, or “spliced out,” in the RNA stage (the intermediary step between DNA and protein synthesis in cells).
The implications of this particular sort of defect are “enormous,” says Pandolfo. People with FA “have the disease because they make too little of a protein, frataxin, but the protein they make is normal.
“So, if one can boost its synthesis, the basic defect in the disease may be corrected.”
In other words, “if we can bypass the problems caused by the repeat expansion,” Bidichandani explains, “the genetic instructions will allow production of a perfectly normal frataxin protein.”
Frataxin’s function
The frataxin protein does its work in mitochondria, the energy-producing “powerhouses” in cells.
Although the protein is present in all tissues of the body, “only certain cells are exquisitely sensitive to frataxin depletion,” says Grazia Isaya, professor of biochemistry and molecular biology and pediatrics at the Mayo Clinic College of Medicine in Rochester, Minn., and a member since 1998 of MDA’s Medical Advisory Committee. These include cells in the heart and pancreas, and large sensory neurons (nerve cells) associated with the spinal cord.
“In fact, even in the central and peripheral nervous system, there are only certain neuronal cells that are particularly susceptible to frataxin deficiency,” Isaya explains. “Maybe other tissues are sensitive as well, but not to the extent that it becomes clinically obvious — and we do not yet understand why this is the case.”
Isaya cautions that the basic function of the frataxin protein is a polarizing topic among researchers in the field, with some maintaining that frataxin’s function is not yet known.
“My view is the opposite. I think we understand the basic function of the protein, which is to bind iron and make it available to other mitochondrial proteins,” Isaya says. “We certainly don’t understand the details of how it’s accomplished at the molecular and atomic levels, but I think the main function of the protein is well-known.”
What is agreed upon — at least by most — is that in FA, the physical increase in the amount of DNA in the gene for frataxin directly or indirectly results in a decrease in transcription of instructions into RNA and, by extension, decreased levels of frataxin.
Why isn’t there enough frataxin?
The first mechanism posited to explain the decrease in frataxin transcription was proposed by Massimo Pandolfo and colleagues when they initially identified the GAA expansion.
The group theorized that the repeats would form an abnormal “triplex” DNA structure, one variant of which was dubbed “sticky DNA.”
Such abnormal structures have been shown in bacterial and various mammalian cell culture models to reduce transcription.
“So these early data showed that the expanded GAA repeat was capable of reducing frataxin RNA production,” Bidichandani says. “In other words, no other factors, such as epigenetic modification [a sort of biochemical gene regulation mechanism], are necessary.”
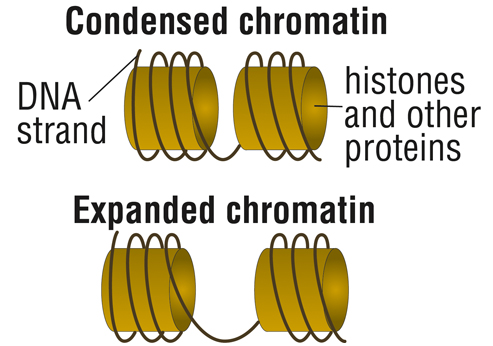
More recent work indicates that the GAA expansion also regulates activity of the frataxin gene through epigenetic controls that causegene silencing.
In the case of FA, this involves compacting chromatin, the structure formed by DNA and the proteins that DNA is wrapped around, called histones.
With such “condensed” chromatin, DNA is closed, preventing the cell’s protein-building machinery to access the genetic instructions.
“With the epigenetic story, you get a closed chromatin formation near the expanded repeat sequence,” Bidichandani explains. “We also have new evidence that this might be occurring near the area of the gene where transcription starts as well.”
The two mechanisms — abnormal DNA structure and epigenetic gene silencing — aren’t mutually exclusive and may in fact overlap.
In any case, decrease of frataxin RNA leads to decrease in frataxin protein production, which in turn leads to abnormal iron overload and metabolism in the mitochondria.
Iron overload
Over the last several years, MDA grantee Des Richardson and colleagues at the University of Sydney, Australia, have focused on pinpointing the pathways that lead to iron accumulation.
In 2008, the group published study results describing the downregulation of three iron-trafficking pathways caused by decreased frataxin production: mitochondrial iron storage, iron-sulfur cluster (ISC) synthesis and heme synthesis.
ISCs and heme are important molecules that drive a number of vital cellular processes affecting every compartment and a number of different pathways in the cell.
“Normally, iron comes into the mitochondria, and it’s utilized very efficiently for the synthesis of iron-sulfur clusters and heme,” Richardson explains. “It comes in, it incorporates into the ISCs, incorporates into the heme, and then very quickly these complexes are shuttled out, back into the cell.”
Without frataxin to handle iron safely and efficiently, mitochondria are no longer able to generate the ISCs and heme. When the cell detects the problem, it senses it as a situation of iron insufficiency and works to fix it by bringing more iron into the cell.
On its surface, the cell increases the levels of a molecule called the transferrin receptor, which brings in more iron from a protein in the blood called transferrin.
The iron is targeted to the mitochondria, but when it gets there it’s unable to be used. It accumulates and the mitochondria become iron-loaded at the same time other areas of the cell exhibit iron deficiency.
“It’s a paradoxical situation,” Isaya says. “The cells feel that they are depleted of iron, when in fact they’re accumulating it in the mitochondria.”
Oxidative stress
In addition to the disruption of ISC and heme generation, and inappropriate iron levels, the problem of iron overloading in the mitochondria can lead to extensive cellular damage via oxidative stress.
Because the mitochondria are unable to direct the binding of iron with proteins to make the complexes necessary for ISC or heme synthesis, iron is left “free,” able to induce the conversion of energy-production byproducts into toxic chemicals called free radicals.
When produced in excess, or when cells become unable to detoxify them, free radicals become dangerous, causing extensive damage to cellular structures, including DNA, and eventually killing the cell.
Among the variety of outcomes associated with iron-triggered free radical activity are heart disease and neurological problems.
An understanding of free radical damage led to some of the first treatment strategies for FA.
FA treatment strategy: Antioxidants
Antioxidants are drugs designed to offer some protection against free radical damage in cells and to enhance mitochondrial energy production by rendering free radicals harmless.
Coenzyme Q10, vitamin E and idebenone were among the first antioxidant compounds to be tested in FA after the frataxin gene discovery. These studies, particularly of idebenone, continue today. A long history of clinical trials of the drug has produced some intriguing results. (See Idebenone Clinical Trials.)
Idebenone (under the brand name Catena) is not approved for use in the United States. It did receive conditional market approval for FA treatment in Canada in July 2008. Around that same time, however, it failed to obtain marketing approval in Europe, where its trade name is Sovrima.
Although antioxidants alone can’t cure FA, this class of drug may provide some benefit, alone or in combination, with other therapies.
FA treatment strategy: Iron chelation
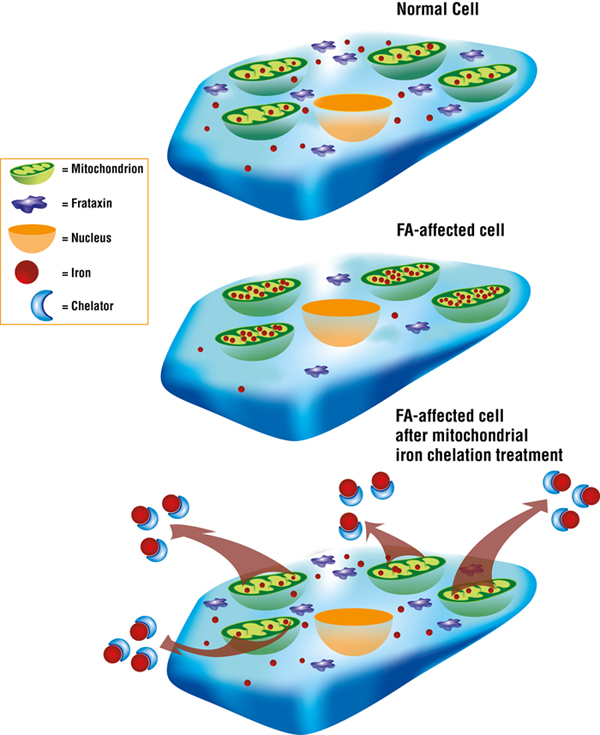
Des Richardson and colleagues have done extensive work on iron chelation, a therapeutic strategy that aims to target toxic mitochondrial iron and remove it via compounds called iron chelators.
In July 2008, Richardson’s team published study results showing that treatment with iron chelation significantly decreased iron levels in the heart, and limited the harmful increase in the size of the cardiac muscle (a condition called myocardial hypertrophy) in mice that don’t express frataxin in the heart.
Precision in iron chelation therapy is crucial.
“You have to be very careful — you’re on the knife’s edge,” Richardson explains. “You have to give an appropriate dose that potentially will remove the iron from the mitochondria without causing whole-body iron depletion.”
Although Richardson’s team has demonstrated that iron chelation can improve symptoms in a mouse with an FA-like disease, in the end the animal still gets ill and suffers from heart problems.
“This is because — despite removing the toxic iron — we couldn’t replace the function of the frataxin, which is essential for the synthesis of iron-sulfur clusters and heme,” Richardson says.
The role for iron chelation as a treatment for FA is thus limited.
Results from an ongoing clinical trial with study sites in Belgium, France, Italy and Spain may increase understanding of how iron chelation may (or may not) fit into an FA therapy regimen.
The study aims to assess the tolerability and safety of the iron chelation drug deferiprone in people with FA. In development by the pharmaceutical company ApoPharma, headquartered in Canada, the drug has been licensed for treatment in Europe and Asia of the iron-loading disorder thalassemia.
“I’m cautious but hopeful that iron chelators might be used to reduce the amount of toxic mitochondrial iron, but again it will not fix the problem — that is the lack of frataxin,” Richardson says.
“Eventually we have to generate a technology that will restore or replace frataxin function, and that’s really the ‘Holy Grail.’”
FA treatment strategy: Erythropoietin
Erythropoietin, or EPO, is a sugar-coated protein, or glycoprotein, secreted by the kidneys.
In addition to its neuro- and cardioprotective properties, EPO:
- stimulates production of red blood cells;
- works as a powerful growth factor that can increase cell size and numbers of mitochondria; and
- increases frataxin protein production through an as-yet-undetermined mechanism.
Limitations to prolonged EPO treatment include harmful side effects such as increased blood-cell production and the risk of tumor growth.
In 2005, a group of Austrian investigators targeted human erythropoietin to cultured lymphocytes (a type of white blood cell) taken from people with FA, and found the cells significantly increased production of frataxin protein.
The researchers also studied people with end-stage kidney disease who were being treated with EPO, and found a significant increase in frataxin production in those patients within 48 hours of treatment.
In 2010, another Austrian research team (including some of the same researchers from the 2005 study) reported results of a small “proof-of-concept” trial in which 12 people with FA received EPO injections three times a week for eight weeks.
Eight of the 10 trial participants exhibited a stable increase of frataxin levels over the study period, with individual increases ranging from 15 to 63 percent.
Ataxia severity, as measured by the assessment and rating of ataxia (SARA) scale, showed a 6 percent improvement in study participants, and measures of oxidative stress were significantly reduced.
In an open-label follow-up study, eight of the 10 participants from the first trial underwent a six-month extended treatment phase. Results included significant clinical improvement based on measurements from two different ataxia rating scales, with changes in locomotion and speech contributing the most to overall score improvement. Parameters indicative of oxidative stress also were markedly reduced after treatment.
Finally, building on their earlier studies, several members of the Austrian group, with colleagues, showed that a derivative of EPO called carbamylated erythropoietin (CEPO) increases frataxin levels in FA-affected cells, but does not, as EPO does, stimulate increased blood-cell production. This makes it a potential alternative for treatment of FA.
FA treatment strategy: HDAC inhibitors
Using a class of drugs called histone deacetylase (HDAC) inhibitors, scientists hope to “turn back on” the frataxin gene, allowing cellular machinery to read its genetic instructions and produce the frataxin protein.
Early attempts with a synthetic small molecule reversed the deleterious effects of the GAA repeat expansion and turned the frataxin gene back on to a small extent in cell culture studies. In further testing, however, in Massimo Pandolfo’s mouse model for FA, it was found that the molecule didn’t get into the brain.
“That got me thinking that the mechanism of gene silencing may have something to do with chromatin and epigenetics,” says Joel Gottesfeld, professor in the department of molecular biology at Scripps Research Institute in La Jolla, Calif.
“And that led me to propose that molecules that could revert inactive chromatin to active chromatin might be of benefit.”
Gottesfeld’s lab began a search for HDAC inhibitors that could turn the frataxin gene back on, and found one, a molecule called BML210. A chemist in the lab synthesized a collection of derivatives of the compound, eventually leading up to a molecule called 106.
The 106 molecule was passed on to Pandolfo’s lab, where it was shown that it does activate the frataxin gene in both the brain and heart of their FA mouse model, and subsequently leads to increased protein levels.
After a number of preliminary experiments to show the molecule wasn’t toxic even in prolonged treatment situations, Gottesfeld and Scripps filed a provisional patent application and began looking for a commercial partner to help move the drug forward.
A number of companies expressed interest, Gottesfeld says, and in April 2007, Repligen Corp. of Waltham, Mass., entered into a commercial license with Scripps for exclusive rights to further develop the compound.
“Repligen made Scripps the best offer — not just financially, but in terms of our confidence in them sincerely wanting to take this forward,” Gottesfeld says.
“That’s even more important than the dollars that the company may pay to Scripps for the rights, because it’s of no use unless the company really is dedicated to do the work.”
James Rusche, Repligen senior vice president of research and development, says, “We are really excited about this approach. It’s not treating a symptom — it actually treats the fundamental basis of the disease.”
Repligen has since synthesized hundreds of derivatives of 106, one of which, RG2833, has been designated for clinical development.
Studies have confirmed that RG2833 is most potent against HDAC3, an enzyme that, when blocked, increases frataxin production.
“We think this is a sound and exciting opportunity to develop a treatment for FA,” Rusche says. “Small molecule drug development for a neurodegenerative disease is difficult, but with support from academic and patient advocacy groups we feel Friedreich’s ataxia is the right place to try.”
MDA provided Repligen with two grants to fund the early stages of the RG2833 program. The first, awarded in late 2007, totaled nearly $1 million. In December 2009, MDA awarded a new grant to the company totaling $731,534, to support the ongoing development of RG2833, including completion of the preclinical testing required to move the experimental drug toward human clinical trials.
Repligen has filed an Investigational New Drug application with the U.S. Food and Drug Administration (FDA) for a phase 1 study of RG2833. Pending approval by the FDA, the study, which will enroll up to 40 healthy volunteers without FA, will evaluate the safety and pharmacokinetic profile (the way the drug behaves in the body) of the compound.
Researchers also will evaluate biochemical indicators called biomarkers in the participants’ blood as a means of measuring RG2833’s activity.
This study will be the first step in the clinical development of RG2833 as a potential treatment for FA.
The FDA has granted RG2833 orphan drug designation, which may qualify Repligen to receive seven years of market exclusivity in the United States, provided that it is the first company to receive approval for RG2833 for FA.
Combining the use of different treatment strategies is likely, says Gottesfeld.
“A treatment for Friedreich’s ataxia may turn out to be a combination of treating symptoms with either an iron chelator and/or an antioxidant, plus boosting levels of frataxin protein — either by protein replacement or by our means, turning the gene back on.”
FA treatment strategy: Gene therapy
Various gene therapy-based strategies also hold the potential to benefit individuals with FA. Although it’s relatively early in the game, a limited number of studies in mice and human cells have yielded some encouraging results.
In a 2005 study, researchers used lentiviral or adeno-associated viral (AAV) vectors to carry the human frataxin gene intofibroblasts (cells that mature into a variety of connective tissue types) from FA patients. (A vector is a delivery vehicle for therapeutic genes.)
Results included increased frataxin protein levels and a reduction in the treated cells’ sensitivity to oxidative stress.
A 2007 study analyzed the feasibility of gene insertion via the emptied-out shell of the herpes simplex virus type 1 (HSV-1), partnered with a stretch of DNA called an amplicon that helps the vector target a specific cell type.
Results in FA-affected mice and human cells showed highly efficient DNA transfer and increased levels of frataxin production.
Similar positive results in 2007 came from a Spanish study in which mice were engineered so that researchers could eliminate, or “knock out,” frataxin activity specifically in motor neurons.
The mice developed neurological symptoms after four weeks, but achieved full recovery in as few as four weeks after receiving injections of HSV-1 amplicon vectors carrying DNA that codes for human frataxin.
A Spanish study published in 2010 describes the successful use of artificial chromosomes and similar constructs called episomesto act as vectors for transporting genes to their targets.
R. Mark Payne, a professor of pediatrics and a pediatric cardiologist at the Indiana University School of Medicine in Indianapolis, is working on gene replacement therapy for FA.
His strategy involves using a construct called TAT-frataxin fusion proteins as a mechanism for replacing the missing FA gene product.
(For an interview with R. Mark Payne about heart problems in FA, see Treating the FA-Affected Heart.)
Where are we now?
The FA disease process is extremely complex. Scientists still are refining their knowledge of its causes and effects, but a great deal of progress has been made. Still, more needs to be done.
It’s becoming clear that in addition to mitochondrial consequences, there are other far-ranging cellular consequences. A number of pathways that are normally active in cells are lost in the absence of frataxin and their loss then contributes to the progression of FA symptoms, Grazia Isaya notes.
“This may explain why single drugs like antioxidants or iron chelators alone cannot be entirely successful in terms of improving the disease in patients.
“Perhaps only by increasing the levels of frataxin can one ultimately treat the disease.”
To see a listing of current and completed clinical trials in FA, go to ClinicalTrials.gov and enter “Friedreich’s ataxia” in the search box.
The entire In Focus: Friedreich’s Ataxia report can be seen on the Friedreich's ataxia disease page.
MDA Resource Center: We’re Here For You
Our trained specialists are here to provide one-on-one support for every part of your journey. Send a message below or call us at 1-833-ASK-MDA1 (1-833-275-6321). If you live outside the U.S., we may be able to connect you to muscular dystrophy groups in your area, but MDA programs are only available in the U.S.
Request Information
